
Прогрессирующая мышечная дистрофия Дюшенна
Что такое Прогрессирующая мышечная дистрофия Дюшенна –
Прогрессирующая мышечная дистрофия Дюшенна описана Дюшенном в 1853 г. Частота 3,3 на 100 000 населения, 14 на 100 000 родившихся.
Что провоцирует / Причины Прогрессирующей мышечной дистрофии Дюшенна:
Наследуется по рецессивному, сцепленному с Х-хромосомой типу. В подавляющем большинстве случаев болеют мальчики. Дистрофия Дюшенна связана с поражением гена, ответственного за выработку дистрофина. При обследовании матерей – носителей гена в генетических консультациях (биопсия ворсинок хориона на 8–9-й неделе) выявляют заболевание у мальчиков. Случаи заболевания у девочек крайне редки, хотя и возможны при кариотипе X0, мозаицизме X0/ХХ, X0/ХХХ, X0/ХХХ/ХХХ и при структурных аномалиях хромосом.
Патогенез (что происходит?) во время Прогрессирующей мышечной дистрофии Дюшенна:
Характеризуется перерождением мышечной ткани, замещением ее жировой и соединительной тканью, некрозом отдельных волокон.
Симптомы Прогрессирующей мышечной дистрофии Дюшенна:
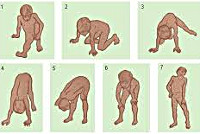
Признаки заболевания проявляются в первые 1–3 года жизни. Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети не усто йчивы, часто спотыкаются, падают. В 2–3 года по являются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке – длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, из положения на корточках или со стула. Вставание происходит поэтапно, с активным использованием рук – «взбирание лесенкой» или «взбирание по самому себе». Атрофии мышц всегда симметричны. Вначале они локализуются в проксимальных группах мышц нижних конечностей – мышцах тазового пояса, бедер, а через 1– 3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей – плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц.
При пальпации мышцы плотны, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Сухожильные рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позднее – рефлексы с двуглавой и трехглавой мышц. Пяточные (ахилловы) рефлексы длительное время остаются сохранными.
Снижение амплитуды осцилляции и увеличение полифазности.
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживаются сужение костно-мозгового канала, истончение коркового слоя длинных диафизов трубчатых костей.
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада нож ек пучка Гиса и др.). Нейроэндокринные нарушения встречаются у 30–50 % больных. Чаще других наблюдаются синдром Иценко– Кушинга, адипозогенитальная дистрофия Бабинского–Фрелиха. Интеллект у многих больных снижен в различной степени.
Течение. Болезнь имеет быстро прогрессирующее злокачественное течение. К 7–10 годам возникают глубокие двигательные расстройства – выраженное изменение походки, снижение мышечной силы, в значительной степени ограничивающие свободное, самостоятельное передвижение больных. К 14–15 годам наступает обездвиженность.
Диагностика Прогрессирующей мышечной дистрофии Дюшенна:
Диагноз ставится на основании данных генеалогического анализа (рецессивный сцепленный с Х-хромосомой тип наследования), клинических обострений болезни (раннее начало в 1–3 года, симметричные атрофии проксимальных групп мышц, развивающиеся в восходящем направлении, псевдогипертрофии икроножных мышц, грубые соматические и нейроэндокринные расстройства, снижение интеллекта, быстрое злокачественное течение болезни), данных биохимических исследований (типично раннее, с 5-го дня жизни ребенка, увеличение активности КФК – в 30–50 раз выше нормы), игольчатой электромиографии и морфологических результатов. позволяющих выявить первично-мышечный (миодистрофический) тип поражения.
Дифференцировать заболевание следует от спинальной амиотрофии Верднига– Гоффманна, рахита, врожденного вывиха бедра.
К каким докторам следует обращаться если у Вас Прогрессирующая мышечная дистрофия Дюшенна:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Прогрессирующей мышечной дистрофии Дюшенна, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна – тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Перспективным направлением является разработка и использование препаратов, нацеленных на восстановление синтеза или замещение дистрофина. В 2020 г. в России был зарегистрирован первый таргетный препарат для лечения мышечной дистрофии Дюшенна – аталурен, который подходит 10% пациентам с нонсенс-мутацией (стоп-кодоном). Препарат позволяет возобновить синтез белка дистрофина в организме, требует пожизненного приема. Другие аналогичные лекарственные средства (этеплирсен, голодирсен) в РФ пока не одобрены к применению. Положительные результаты на стадии клинических испытаний у пациентов с миодистрофией Дюшенна показал микродистрофин. Идет поиск и апробация методов заместительной генной терапии.
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Публикации в СМИ
Мышечная дистрофия Дюшенна — наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением; наследуется по рецессивному X-сцепленному типу. Вариант мышечной дистрофии Дюшенна — мышечная дистрофия Беккера — имеет более доброкачественное течение.
Генетические аспекты • Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна–Беккера, *310200, Xp21.2, ген DMD дистрофина, À рецессивное) — возникает в результате дефектов гена, кодирующего белок дистрофин • Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов • Преобладающий пол — мужской, тем не менее мышечные дистрофии Дюшенна и Беккера могут встречаться у девочек при кариотипе X0, мозаицизмах X0/XX, X0/XXX и структурных аномалиях хромосом.
Патоморфология • Дистрофия мышечных волокон, первично-мышечный тип поражения • Фиброзные изменения в мышечных пучках • Местная воспалительная реакция.
Клиническая картина
• Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно со слабости мышц тазового пояса.
• Уже на первом году жизни отмечают отставание в психомоторном развитии. Больные дети позднее начинают садиться, вставать, ходить.
• Постепенно развиваются слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу утиной. Из горизонтального положения дети встают поэтапно с использованием рук (взбирание лесенкой).
• Отмечаются симметричные атрофии проксимальных групп мышц нижних конечностей (мышцы таза и бедра). Атрофия через 1–3 года распространяется на проксимальные группы мышц верхних конечностей.
• Атрофии мышц приводят к развитию лордоза, крыловидных лопаток, осиной талии.
• Характерна псевдогипертрофия икроножных мышц.
• Мышцы при пальпации плотные, безболезненные.
• Мышечный тонус обычно снижен в проксимальных группах мышц.
• Изменения рефлексов •• Коленные рефлексы исчезают на ранних стадиях заболевания •• Позднее исчезают рефлексы с двуглавой и трёхглавой мышц плеча •• Ахилловы рефлексы обычно длительное время остаются сохранными.
• Дистальная мускулатура конечностей поражается на поздних стадиях заболевания.
• Костно-суставные нарушения — деформации позвоночника, стоп, грудной клетки; рентгенологически обнаруживают сужение костномозгового канала, истончение коркового слоя диафизов длинных трубчатых костей.
• Сердечно-сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность, изменения на ЭКГ.
• Нейроэндокринные нарушения выявляют у 30–50% больных — синдром Иценко–Кушинга, адипозогенитальная дистрофия.
• Психические нарушения — олигофрения в форме дебильности или имбецильности.
• Клинические проявления мышечной дистрофии Беккера обычно начинаются в 10–15 лет. От мышечной дистрофии Дюшенна отличается доброкачественным течением и более поздним возникновением тяжёлых симптомов. Сухожильные рефлексы долгое время остаются сохранными. Поражения внутренних органов менее выражены, интеллект сохранён.
Лабораторные исследования. Для мышечной дистрофии Дюшенна типично раннее (с 5 дня жизни) увеличение активности КФК в крови (в 30–50 раз выше нормы).
Дифференциальная диагностика. Мышечную дистрофию Дюшенна–Беккера дифференцируют от других мышечных дистрофий, рахита, врождённого вывиха бедра.
ЛЕЧЕНИЕ
Режим амбулаторный с наблюдением у невропатолога, хирурга-ортопеда, терапевта и профпатолога, работника социальной сферы и протезиста.
Мероприятия • Лечение мышечной дистрофии Дюшенна направлено на поддержании физической активности пациента и улучшение качества его жизни; как правило, быстро становится неэффективным • Физические упражнения выполняют систематически и по определённой схеме. Короткие перерывы показаны при возникновении болей в мышцах и мышечной усталости • Использование протезов позволяет больным двигаться и замедляет формирование сколиоза • Поддержание дыхания, ИВЛ во время сна для предотвращения синдрома ночной гиповентиляции • Экспериментальные методы, в особенности генная терапия (гены дистрофина и утрофина), чрезвычайно перспективны, хотя и не получили пока клинического распространения.
Оперативное лечение. Ортопедическое вмешательство необходимо при наличии контрактур и фиксации суставов.
Лекарственная терапия • ГК (преднизолон по 0,75 мг/кг/сут) увеличивают мышечную силу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование заболевания • При длительной стероидной терапии необходим тщательный контроль развития побочных эффектов, включающий наблюдение за массой тела, АД, состоянием слизистой оболочки ЖКТ и иммунной системы.
Наблюдение. Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни пациентов.
Течение и прогноз • Течение мышечной дистрофии Дюшенна быстропрогрессирующее, злокачественное • Значительные двигательные расстройства, развивающиеся ко второму десятилетию жизни, ограничивают самостоятельное передвижение больных • Смерть наступает на втором или третьем десятилетии жизни, часто в результате пневмонии • Течение мышечной дистрофии Беккера медленнопрогрессирующее. Больные длительное время сохраняют работоспособность.
Профилактика состоит в генетическом консультировании.
Синонимы • Прогрессирующая мышечная дистрофия Дюшенна • Псевдогипертрофическая мышечная дистрофия Дюшенна • Дистрофия Дюшенна • Болезнь Дюшенна • Миопатия псевдогипертрофическая • Миопатия псевдогипертрофическая Дюшенна.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Примечания • Термин «псевдогипертрофическая прогрессирующая мышечная дистрофия» объединяет мышечные дистрофии Дюшенна и Беккера • Мышечная дистрофия Дюшенна описана в 1853 г. Дюшенном • Мышечная дистрофия Беккера описана в 1955 г. Беккером.
«Ты особенный малыш»: что делать, если у ребенка миодистрофия Дюшенна
Мышечная дистрофия Дюшенна (МДД) – редкое генетическое заболевание, которое обычно поражает мальчиков (примерно одного из 3500–5000). Болезнь развивается из-за мутаций в гене дистрофина и разрушает мышцы: сначала ребенок перестает ходить, а со временем и дышать. Пока заболевание остается неизлечимым.
Эту болезнь врачи в России часто диагностируют поздно. Ольга Гремякова, мама семилетнего Гордея, сама догадалась, что у ее сына миодистрофия Дюшенна: заметила симптомы, нашла информацию и отвезла сына к врачам, которые подтвердили ее опасения.
Ольга и ее семья создали благотворительный фонд «Гордей», чтобы улучшать качество медицинской, социальной и психологической помощи пациентам с МДД и их семьям. В декабре фонд провел первую в России конференцию для родителей и врачей, посвященную миодистрофии Дюшенна.
Поговорили с Ольгой о том, как распознать болезнь и с чем сталкиваются пациенты с МДД и их семьи после постановки диагноза.
От первых симптомов до диагноза
Прислушиваться к себе
Я видела, что Гордей отличается от других ребят, но не понимала, почему. До трех лет он не разговаривал. Был немного неуклюжий, не умел прыгать и бегать. Мне говорили, что причина в тяжелых родах, поэтому нужно немного подождать, и он перерастет эту неуклюжесть. Либо мягко намекали на то, что я просто тревожная мама, которая беспокоится за своего первого малыша. Когда мы проходили обследования перед детским садом, все специалисты в очередной раз сказали, что сын здоров. Не насторожился никто – ни врачи, ни моя мама, доктор медицинских наук.
Кроме того, Гордей был и остается очень требовательным и чувствительным ребенком, с ним сложно взаимодействовать. Родные говорили, что это я воспитала его таким: он капризный и просится на ручки не из-за усталости, а потому что манипулирует. Ну что скажешь в этот момент? Я действительно думала, что сама виновата. Тогда я не знала, что эти особенности тоже связаны с его болезнью.
Заметить симптомы
Я услышала о миодистрофии Дюшенна за пару лет до того, как ее диагностировали у Гордея. Это была история Ольги и Петра Свешниковых – учредителей фонда «Мой Мио», которые усыновили ребенка с МДД. Этот поступок вызвал у меня бесконечные уважение и восхищение. Но тогда я просто прочла об этом и продолжила жить дальше. Спустя какое-то время я увидела в фейсбуке историю папы и сына с МДД. После фразы «я смотрел на его икры, они были такие мускулистые, а сил-то в них не было» картинка в голове полностью сложилась. Я погуглила симптомы и поняла, что с моим ребенком.
Главный признак мышечной дистрофии – прогрессирующая мышечная слабость. Специфические симптомы появляются в разном возрасте и в разных группах мышц, в зависимости от типа мышечной дистрофии. Обычно первые признаки МДД проявляются к двум-трем годам. Ребенок может часто падать, медленно бегать, ходить на цыпочках или вразвалочку. Ему может быть сложно вставать с пола и подниматься по лестнице. Часто у ребенка с МДД непропорционально большие икры. Сначала слабость более выражена в мышцах бедер, но со временем она распространяется и на другие, включая мышцы, участвующие в дыхании. У ребенка может быть задержка двигательного и речевого развития, трудности в обучении. МДД также влияет на память, манеру общения и эмоциональное состояние.
Уже через пару часов Гордей сдал кровь. Как мы дожили до конца дня, я не помню. Ночью проснулась, проверила почту и нашла письмо от лаборатории: уровень креатинкиназы – 23076 при норме у мальчиков до 200!
На следующий вечер я укладывала сына спать и сказала ему: «Гордей, я теперь все про тебя знаю. Знаю, что ты устаешь, что у тебя не очень сильные мышцы, что ты такой вот особенный малыш». Гордюше тогда было четыре с половиной года. Он ничего не сказал, а просто тихонечко заплакал. Наверное, для него было важно, что его наконец понимают.
При подозрении на МДД берут анализ крови на креатинкиназу (КК) – фермент, который мышцы выделяют при повреждении. Высокий уровень КК – характерный признак миодистрофии Дюшенна. Для подтверждения диагноза используют генетические тесты – они находят крупные мутации в гене дистрофина. Если крупных мутаций не нашли, врачи сделают секвенирование (расшифровку последовательности) гена, чтобы найти мелкие точечные мутации.
Если обращать внимание на характерные симптомы, то диагностировать МДД можно рано – например, с года до двух. Но для этого врачи должны заметить признаки миодистрофии Дюшенна в потоке пациентов. По мнению Ольги, идеальным диагностическим решением было бы добавить к неонатальному скринингу тест на креатинкиназу – он простой и стоит около 400 рублей.
К сожалению, примерно половина мальчиков с МДД исходно получает неверный диагноз. Чаще всего гепатит, потому что из-за распада мышц растет уровень ферментов трансаминаз, что обычно соотносят с болезнью печени. Также мальчикам ставят перинатальное поражение ЦНС, аутизм, задержки двигательного и речевого развития и даже ДЦП. Иногда МДД диагностируют очень поздно – в возрасте от 8 до 12 лет, когда симптоматика становится заметной и папы начинают носить мальчиков на руках.
 |
Ольга с детьми. Фото из личного архива
Делать все возможное
Первое изменение в жизни моей семьи – это зашкаливающий уровень боли. Боли от диагноза нашего любимого малыша, которого еще пару дней назад мы считали здоровым. Вскоре у меня развилось состояние предвосхищающего горя: ты знаешь, что ребенок жив, но думая о его диагнозе, понимаешь – все предопределено. Рухнуло все: представление о счастливом материнстве, где я – мама двух прекрасных здоровых малышей, о будущем, где сын будет катать меня на машине и мы вместе будем делать всякие штуки. Рухнула моя идентичность – как мамы и как женщины.
Я плохо помню первые два года жизни после постановки диагноза. Через полгода у меня началась депрессия: не было сил даже на самые обычные дела, я чувствовала себя разбитой, ничто меня не радовало… Мое состояние заметила мама и настояла на обращении к врачу – сама я, несмотря на психологическое образование, депрессии у себя не заметила. Врачи помогли. Все же я не могла пару месяцев лежать лицом к стене – нужно было заниматься детьми.
Мы решили, что будем пользоваться каждым днем и делать для Гордея все возможное, чтобы он чувствовал себя лучше. Да, волшебной таблетки у нас нет, но мы в силах снизить риск контрактур (ограничение подвижности сустава – прим. авт.) голеностопа и других осложнений, которые усаживают мальчиков в коляску раньше.
Физические упражнения, растяжка и низкоинтенсивные аэробные нагрузки (ходьба и плавание) помогают пациентам с МДД сохранить мышечную силу и диапазон движений в суставах. Специальные изделия – туторы – могут поддерживать голеностопный сустав и стопы в правильном положении, замедлять прогрессирование контрактур. Чем дольше ребенок ходит и чем позднее понадобится коляска, тем лучше для сердца и дыхательной системы и тем ниже риск развития сколиоза, требующего оперативного вмешательства.
Чтобы вернуть себе ресурс и сражаться за ребенка, с первого дня учитесь делать ему растяжки ахиллового сухожилия, голеней и бедер – по 20–30 минут ежедневно. Благодаря таким занятиям мальчик сможет ходить дольше. Когда родитель «своими руками» продлевает ребенку активные годы, это очень важно для обоих.
От непонимания до взаимной поддержки
Совершенствовать систему
Со временем мы узнали, как устроена помощь пациентам с миодистрофией Дюшенна. Помимо небольших выплат государство предлагает мальчикам санаторно-курортное лечение. Но пока система плохо понимает, что именно нужно детям с МДД. На опыте Гордея могу сказать, что предлагают обычно не то и не там – хоть и очень стараются. Два раза в год мальчикам и юношам с миодистрофией положена реабилитация – и это не массажи и грязи. Она конечно есть, но далеко не везде соответствует международным стандартам. Более того, не везде она человечная. Иногда мамы с сыновьями сбегают с реабилитации, потому что еда плохая, обращение еще хуже, занятий нет.
Также существует ИПРА – индивидуальный план реабилитации (абилитации). В него вписывают технические устройства, предлагаемые государством, например коляски и туторы. Но часто эти устройства не подходят ребенку, и родители вынуждены покупать другие за свой счет – при этом стоимость компенсируют не всегда. Это очень чувствительный вопрос для семьи, и самые большие трудности возникают именно со средствами передвижения для неходячих мальчиков.
Получается, что государство помогает, но сами механизмы негибкие и нуждаются в пересмотре. По международным стандартам реабилитации ребятам с МДД нужны теплый бассейн, правильные растяжки, полный медицинский чекап и сильная мультидисциплинарная команда. Еще год назад в России не было центров, где могут все это предложить. Сейчас самый современный вариант – реабилитация в Центральной клинической больнице, программу которой разработали эксперты ЦКБ в сотрудничестве с нашим фондом. Она соответствует лучшим международным практикам и в этом смысле уникальна для России.
Но такой центр один, а мальчики с МДД рождаются по всей стране. Их примерно четыре тысячи, при этом для системы здравоохранения видны не более полутора тысяч. В любом случае, наших мальчиков слишком много для того, чтобы всех отправлять в Москву. Ребятам нужна реабилитация в региональных центрах, ведь многим из них сложно передвигаться. Поэтому наша задача – создавать такие центры и обучать специалистов на местах. Для этого нам нужно сильное пациентское сообщество и некоммерческие организации, которые будут влиять на неповоротливую государственную систему и улучшать ее работу.
Отстаивать интересы
Мы стремимся к тому, чтобы в России приняли клинические рекомендации по ведению пациентов с МДД. Хотим, чтобы мальчики и их семьи получали все виды помощи – медицинскую, социальную и психологическую, и чтобы эта помощь соответствовала международным стандартам.
Есть и другие организации, занимающиеся миодистрофией Дюшенна – фонд «Мой Мио» (6 лет) и Родительский проект (3 года). У каждой из них более 500 подопечных. Они реализуют медицинские и образовательные программы для пациентов с МДД, оказывают информационную, консультативную и адресную помощь семьям.
После постановки диагноза семьи сталкиваются с дефицитом информации в отношении препаратов, физической терапии, пищевых добавок и приема стероидов. Именно потребность узнать больше помогает родителям разобраться в разных аспектах заболевания, а значит – принимать лучшие решения в интересах своего ребенка и сохранять чувство контроля.
Еще один способ вернуть контроль – присоединиться к родительскому сообществу. Помимо информации в нем можно найти опору, общаясь с другими родителями. У фонда есть сообщество «ProДюшенн», в котором более четырехсот участников, среди которых родители и врачи. Все наши семьи разные: у одних маленькие мальчики с МДД, у других юноши, а есть семьи, где сыновья уже ушли. Но цель у всех одна – поддерживать друг друга и отстаивать интересы пациентов с Дюшенном при взаимодействии с государством.
Также для нас важно, чтобы мамы мальчиков с МДД и их дочери могли выбирать свое репродуктивное будущее и знали об этом. Болезнь развивается из-за мутаций, которые в 70% случаев возникают в Х-хромосоме матери и передаются сыновьям. Если в семье уже есть мальчик с МДД, другие дети также могут унаследовать мутацию. Но предимплантационная диагностика и ЭКО позволяют планировать рождение здоровых детей после появления малыша с МДД. И если я решусь на третьего ребенка, то, скорее всего, обращусь к этим методам. Девочкам, чьи мамы – носительницы мутаций в гене дистрофина, можно сделать анализ ДНК и проверить, передалась ли мутация.
 |
Команда фонда «Гордей». Фото из личного архива
Справиться с кризисом
При МДД родители сталкиваются со множеством кризисных ситуаций. Часто травматичной становится сама постановка диагноза, когда результат генетического теста присылают по почте, а врачи отвечают на звонки без особого желания. Мальчик может столкнуться с переломом и потом долго восстанавливаться, либо теряет способность ходить. Он может оказаться в кресле и без перелома, когда происходит срыв компенсации – снижение мышечной силы. Молодому человеку может потребоваться респираторная поддержка, и это тоже может быть кризисным моментом – как и любое изменение в жизни, указывающее на переход болезни в более тяжелую стадию. И конечно, в таких ситуациях лучше работать с психологом индивидуально.
Еще есть переходный возраст. Это сложное время для любой семьи, но особенно для родителей мальчиков с МДД. Если ребенок принимает кортикостероиды, он не очень сильно растет – а расти ему важно. Для этого придется снизить дозу гормонов, но тогда он начнет слабеть. И этот момент пересмотра лечения, отношений с ребенком и его личных границ тоже бывает очень непростым.
У ребенка с МДД могут быть трудности с учебой, агрессивное поведение, тревожное расстройство или депрессия. Если знать об этом и вовремя обратить внимание, многие вещи можно скорректировать и улучшить качество жизни ребенка и родителей.
Недавно фонд запустил проект психологической поддержки родителей «Передышка». Мы провели уже три встречи, в начале следующего года надеемся параллельно запустить «Передышку» для пап, где будет психолог-мужчина, а также индивидуальную психологическую поддержку.
Искать лечение
Создать универсальное лекарство для МДД сложно. Во-первых, ген дистрофина огромный, и в нем может быть очень много разных мутаций – известно уже около 10 тысяч. Во-вторых, лекарства доставляют в клетки с помощью вирусных векторов (генетических конструкций – прим.авт.), у которых есть определенная емкость. Можно поместить в вирус только фрагмент гена дистрофина. Кроме того, у части пациентов имеется предсуществующий иммунитет к самим вирусам. В-третьих, чтобы лекарство проникло в мышцы, оно должно попасть в общий кровоток – а значит, пройти через печень, которая постарается удалить его из организма. Возникает конфликт: врачи пытаются доставить лекарство в мышцы, а тело сопротивляется и пытается его вывести.
Но таргетные препараты уже появились – и это большая подвижка. Со временем должно появится лекарство, которое подойдет многим пациентам с МДД. Значит нам нужно выявлять болезнь как можно раньше, чтобы быстрее начинать лечение.
24 ноября 2020 года в России зарегистрировали первый таргетный препарат для лечения МДД – аталурен. Он подходит 13% пациентов с МДД, имеющим нонсенс-мутации – точечные замены в гене дистрофина. По словам производителя, лекарство помогает восстанавливать уровень дистрофина и замедлять прогрессирование болезни. Существуют и другие препараты – вайондис, вилтепсо и экзондис, но они также предназначены для пациентов с определенными мутациями.
Традиционное лечение МДД направлено на устранение симптомов. Для повышения мышечной силы и продления двигательной активности врачи назначают пациенту кортикостероиды, но они увеличивают риск переломов и со временем могут привести к набору веса и повышению давления. Для сохранения функций сердечной мышцы применяют ингибиторы ангиотензинпревращающего фермента (АПФ) или бета-блокаторы.
Найти общий язык
Сейчас Гордей учится в нулевом классе. Он чувствует, что справляется, и совершенно счастлив. У него есть друзья, его хвалят учителя. Гордей учится играть в шахматы, с удовольствием изучает испанский и осваивает скетчинг. Для него и других ребят с МДД очень важны вопросы образования, социальной среды и общения.
Так же важна реакция врача на ребенка с миодистрофией. Если врач общается с ним тепло, уважительно и без страха – это одна история. Но когда врачи боятся диагноза, избегают общения с ребенком и ведут себя скованно – и родители, и сами дети это чувствуют. И то, как реагируют на ребенка врачи, учителя, чиновники на комиссиях по инвалидности – это вовсе не «сходил и забыл». Чувствуется негласная стигма «идите умирайте дома, лекарства нет» – и мы должны с ней бороться. Во-первых, лекарства появляются, их будет больше, они будут лучше. Во-вторых, в современном обществе никто не должен быть оставлен без помощи. Для этого нужно учиться делать все вместе – жить, работать, решать гражданские дела. И в этом смысле не может быть и речи о том, чтобы закрыться и угасать дома.
Сохранить веру
Нам всем нужна вера, и ее очень сложно сохранить. Особенно уязвимы родители взрослых сыновей. Ведь Дюшенн – одно из самых травмирующих заболеваний, причем травмируется вся семья, но особенно – мама, которая, как правило, осуществляет основной уход за сыном. Мамы сильно изранены, многим нужна психологическая поддержка и помощь в уходе за ребенком.
Бывает, что родители молодых людей говорят родителям мальчиков помладше: посмотрим, что вы скажете через 10 лет. Да, они испытали то, через что пока не прошла я. И тогда я думаю – действительно, а что я скажу через 10 лет? Я не знаю, я ли это буду или уже не я. Смогу ли сохранить себя, свою любовь, своего ребенка? Я делаю все, что могу, как и моя семья. Боюсь ли я будущего? Я надеюсь, что справлюсь. Ну а там – посмотрим, что я скажу через 10 лет.
Мышечная Дистрофия: Причины, Симптомы и Лечение
Наиболее распространенная форма мышечной дистрофии — мышечная дистрофия Дюшенна — обычно поражает мальчиков, но некоторые формы могут проявиться и у взрослых.
В настоящее время мышечная дистрофия неизлечима, но некоторые физические и медицинские процедуры могут облегчить симптомы и замедлить прогрессирование болезни.
В этой статье мы рассмотрим диагностику, симптомы и лечение мышечной дистрофии, а также рассмотрим текущие исследования будущих методов лечения.
Содержание:
- Что такое мышечная дистрофия?
- Симптомы
- Причины
- Диагностика
- Лечение
- Текущие исследования
Короткие факты о мышечной дистрофии
Вот некоторые ключевые моменты о мышечной дистрофии. Более подробная и вспомогательная информация приведены в основной статье.
- Мышечная дистрофия — это совокупность заболеваний, приводящих к атрофии мышц
- Мышечная дистрофия Дюшенна является наиболее распространенным типом
- Отсутствие белка, называемого дистрофином, является основной причиной мышечной дистрофии
- В настоящее время проводятся испытания генной терапии для борьбы с болезнью
- В настоящее время нет возможности излечиться от мышечной дистрофии
Что такое мышечная дистрофия?

Мышечная дистрофия приводит к постепенному ослабеванию скелетной мускулатуры
Мышечная дистрофия — это заболевание, поражающее мышцы, наиболее распространенные формы которого могут затрагивать 1 на каждые 5000 мужчин.
Заболевание обусловлено генетическими мутациями, которые мешают производству мышечных белков, необходимых для создания и поддержания здоровых мышц.
Болезнь генетическая, и, следовательно, история мышечной дистрофии в семье увеличивает вероятность развития заболевания у человека.
Существует несколько типов мышечной дистрофии, в том числе:
- Мышечная дистрофия Дюшенна — самая распространенная форма болезни. Симптомы обычно начинаются до третьего года ребенка; они, как правило, уже привязаны к инвалидному креслу к 12 и умирают от дыхательной недостаточности в 20-25 лет.
- Мышечная дистрофия Беккера — подобные симптомы к Дюшенна, но с более поздним началом и медленной прогрессией; смерть обычно наступает к сорока-пяти годам.
- Миотоническая (болезнь Штейнтера) — миотоническая форма — наиболее распространенная форма с началом у взрослых. Она характеризуется неспособностью расслабить мышцу после сокращения. Часто сначала поражаются мышцы лица и шеи. Симптомы также включают катаракту, сонливость и аритмию.
- Врожденный — этот тип может быть очевиден с рождения или до достижения двухлетнего возраста. Он затрагивает девочек и мальчиков. Некоторые формы прогрессируют медленно, тогда как другие могут быстро перемещаться на новые группы мышц и вызывать значительные изменения.
- Плече-лопаточно-лицевая — начаться может быть почти в любом возрасте, но чаще всего наблюдается у подростков. Мышечная слабость часто начинается с лица и плеч. Люди с этим типом могут спать, со слегка открытыми глазами и не могут полностью закрывать веки. Когда человек поднимает руки, то лопатки выступают как крылья.
- Пояса конечностей (Эрба) — этот вариант начинается в детстве или подростковом возрасте и сначала проявляется на мышцах плеча и бедра. У больных мышечной дистрофией этого типа может возникнуть проблема с поднятием передней части стопы, что приводит к частым спотыканиям.
- Окулофарингеальная мышечная дистрофия — начало между 40 и 70 годами. Сначала затрагиваются веки, горло и лицо, за которыми следуют плечо и таз.
Симптомы мышечной дистрофии
Ниже приведены симптомы мышечной дистрофии Дюшенна, наиболее распространенной формы заболевания. Симптомы мышечной дистрофии Беккера сходны, но она имеет тенденцию к началу в 20-25 лет или позже, более мягкая и прогрессирует медленнее.
Ранние симптомы
- Ковыляющая походка
- Боль и скованность мышц
- Сложность при беге и прыжках
- Хождение на носочках
- Трудность сидеть или стоять
- Сложности обучения, такие как развитие речи позже обычного
Поздние симптомы
- Неспособность ходить
- Сокращение длины мышц и сухожилий с дальнейшим ограничением движения
- Проблемы с дыханием могут стать настолько серьезными, что необходима медицинская помощь
- Может проявиться сильное искривление позвоночника если мышцы недостаточно сильны, чтобы поддерживать его структуру
- Мускулы сердца могут быть ослаблены, что приводит к сердечным проблемам
- Трудность глотания — это может вызвать аспирационную пневмонию, и иногда необходима питательная трубка
Причины мышечной дистрофии
Мышечная дистрофия вызвана мутациями на Х-хромосоме. Каждый тип мышечной дистрофии обусловлен различным набором мутаций, но все они предотвращают образование организмом дистрофина. Дистрофин — это белок, необходимый для построения и восстановления мышц.
Мышечная дистрофия Дюшенна вызвана специфическими мутациями в гене, который кодирует цистоскелетный белок дистрофин. Дистрофин составляет всего 0,002 процента от общего количества белков в поперечно-полосатых мышцах, но это незаменимая молекула для общего функционирования мышц.
Дистрофин является частью невероятно сложной группы белков, которые позволяют мышцам работать правильно. Белок помогает скреплять различные компоненты внутри мышечных клеток вместе и связывает их всех с сарколеммой — наружной мембраной.
Если дистрофин отсутствует или деформирован, этот процесс работает некорректно, и нарушения происходят во внешней мембране. Это ослабляет мышцы и может также активно разрушать сами мышечные клетки.
При мышечной дистрофии Дюшенна дистрофин почти полностью отсутствует; чем меньше дистрофина, тем хуже симптомы и этиология заболевания. При мышечной дистрофии Беккера происходит уменьшение количества или размера белка дистрофина.
Ген, кодирующий дистрофин, является самым большим известным геном у людей. Более 1000 мутаций в этом гене были идентифицированы при мышечной дистрофии Дюшенна и Беккера.
Диагностика мышечной дистрофии
Существуют различные методы, используемые для окончательной диагностики мышечной дистрофии:
- Ферментный анализ — поврежденные мышцы вырабатывают креатинкиназу (КК). Повышенные уровни КК в отсутствие других видов мышечного повреждения могут указывать на мышечную дистрофию.
- Генетическое тестирование — поскольку известно что генетические мутации происходят при мышечной дистрофии, эти изменения могут быть обнаружены.
- Сердечный мониторинг — электрокардиография и эхокардиограммы могут помочь при обнаружении изменений в мускулатуре сердца. Это особенно полезно для диагностики миотонической мышечной дистрофии.
- Мониторинг легких — проверка функции легких может дать дополнительные доказательства.
- Электромиография — игла помещается в мышцу для измерения электрической активности. Результаты могут показать признаки мышечной болезни.
- Биопсия — удаление части мышцы и исследование ее под микроскопом может показать признаки развития мышечной дистрофии.
Лечение мышечной дистрофии
В настоящее время излечение от мышечной дистрофии невозможно. Лекарства и различные методы лечения замедляют прогрессирование заболевания и поддерживают мобильность пациента в течение максимально длительного времени.
Препараты
Две наиболее часто назначаемые группы препаратов для мышечной дистрофии:
- Кортикостероиды — хотя этот вид лекарств может помочь увеличить мышечную силу и замедлить прогрессирование, их долгосрочное использование может ослабить кости и увеличить прибавку в весе
- Сердечные препараты — если мышечная дистрофия влияет на нормальную деятельность сердца, могут быть полезны бета-блокаторы и ингибиторы ангиотензинпревращающего фермента (АПФ)
Физиотерапия
- Общие упражнения — ряд упражнений по движению и растяжке может помочь бороться с неизбежным внутренним сдвижением конечностей по мере сокращения длины мышц и сухожилий. Конечности, как правило, остаются в фиксированной позиции, а эти упражнения могут помочь передвигаться дольше. Стандартные аэробные упражнения с низким уровнем воздействия, такие как ходьба и плавание, также могут помочь замедлить прогрессирование болезни.
- Дыхательная помощь — поскольку мышцы, используемые для дыхания, становятся слабее, может потребоваться использование устройств, которые помогут улучшить доставку кислорода ночью. В наиболее тяжелых случаях пациенту, возможно, потребуется использовать аппарат для искусственного дыхания.
- Средства передвижения — трости, инвалидные коляски и ходунки.
- Ортезы — они удерживают мышцы и сухожилия растянутыми и помогают замедлить их укорочение. Они также дают дополнительную поддержку пациенту при перемещении.
Текущие исследования мышечной дистрофии
Многое известно о механизмах развития мышечной дистрофии, как мышечном, так и генетическом, и хотя полное излечение еще за горизонтом, есть пути исследований, которые приближаются все ближе и ближе к лекарству.
Генная заместительная терапия

Генная терапия — всего лишь один из путей исследования в области лечения мышечной дистрофии.
Поскольку обнаружен специфический ген, участвующий в развитии мышечной дистрофии, замещающий ген, который сможет создавать недостающий белок дистрофин, является разумным решением.
Но с этим подходом возникают сложные проблемы, в том числе потенциал иммунной системы для отторжения нового белка и большой размер гена дистрофина, который необходимо заменить. Существуют также трудности с нацеливанием вирусных векторов непосредственно на скелетную мышцу.
Другой подход нацелен на производство утрофинов. Утрофин — это белок, подобный дистрофину, который не зависит от мышечной дистрофии. Если бы производство утрофина могло быть усилено, болезнь была бы остановлена или замедлена.
Изменение производства белка
Если ген дистрофина считывается с помощью механизмов синтеза белка и достигается мутация, то он останавливается и белок остается незавершенным. Сейчас испытываются препараты, которые заставляют белково-производственное оборудование клетки пропускать мутированное содержимое и продолжать создавать дистрофин.
Препараты для отсрочки мышечной атрофии
Вместо того, чтобы нацеливаться на гены стоящие за мышечной дистрофией, некоторые исследователи пытаются замедлить неизбежное истощение мышц.
Мышцы, в стандартных обстоятельствах, могут сами по себе регенерироваться. Исследования по контролю или увеличению этих восстановительных процессов могут открыть некоторые новые возможности для людей с мышечной дистрофией.
Исследования стволовых клеток
Исследователи рассматривают возможность введения мышечных стволовых клеток, способных продуцировать недостающий белок дистрофин.
Текущие проекты рассматривают наиболее полезные типы клеток для использования и способы их доставки в скелетные мышцы.
Трансплантация миобластов
На ранних стадиях мышечной дистрофии миобласты (также называемые сателлитными клетками) восстанавливают и заменяют дефектные мышечные волокна. Когда миобласты истощаются, мышцы медленно превращаются в соединительную ткань.
В некоторых исследованиях были предприняты попытки ввести модифицированные клетки миобласта в мышцы, чтобы они заняли место истощенных природных миобластов.
Мышечная дистрофия, или миодистрофия
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Мышечная дистрофия: причины появления, симптомы, диагностика и способы лечения.
Определение
Мышечные дистрофии – это большая группа наследственных заболеваний, которые характеризуются прогрессирующей слабостью и дегенерацией скелетных мышц, то есть потерей мышечной массы. К наиболее распространенным миодистрофиям относятся миодистрофия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия), мышечная дистрофия Дюшенна и дистрофия Беккера. Также в эту группу входят дистрофия Эмери-Дрейфуса, миодистрофия Эрба-Рота и другие.

Миодистрофия Дюшенна поражает в основном мальчиков, а распространенность этого заболевания составляет 3,3 на 100 000 населения, 1 из 3500 новорожденных мальчиков страдает данной патологией. Этот вид дистрофии часто выделяют в одну группу с миодистрофией Беккера (частота встречаемости 1 на 20 000 новорожденных). Миодистрофии Дюшенна и Беккера наследуются по Х-сцепленному рецессивному типу. Это означает, что повреждение находится в Х-половой хромосоме и передается от матери к сыну, а дочери являются носительницами и, как правило, сами не болеют.
Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей.
Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них – Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
Частота встречаемости миодистрофии Эрба-Рота составляет от 1,5 до 2,5 случаев на 100 тыс. населения. Этому типу мышечной дистрофии подвержены и мальчики, и девочки. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно, то есть патология проявляется, если ребенок получает аномальный ген от каждого из родителей. Каждый родитель может быть носителем дефектного гена, но обычно остается здоровым. Около 30% генных мутаций возникают de novo, то есть не наследуются, а появляются «ниоткуда» и далее могут передаваться потомству.
Причины появления мышечной дистрофии
Причиной развития разных миодистрофий являются патологии в генах – известно порядка 25 генов, ответственных за развитие врожденных миодистрофий.
При мышечной дистрофии Дюшенна вследствие мутации нарушается выработка белка дистрофина, который обеспечивает прочность, стабильность и функциональность мышечных волокон, и его нехватка приводит к повреждению мембран мышечных клеток (миоцитов).
Классификация заболеваний
Мышечные дистрофии могут классифицироваться в зависимости от того, какой белок подвергся мутации. Кроме того, их подразделяют по типу наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные.
Симптомы мышечной дистрофии
Мышечная дистрофия Дюшенна обычно манифестирует в возрасте 2-3 лет. Патологические процессы сначала происходят в мышцах ног, дети могут ходить на пальцах, вразвалку, отмечается избыточное выгибание позвоночника вперед – лордоз. Детям становится сложно бегать, прыгать, подниматься по лестнице, вставать с пола. Для разных типов миодистрофий характерным является симптом Говерса – вследствие слабости мышц бедер и тазового пояса больному, чтобы подняться из положения на корточках, приходится опираться руками об пол, затем подниматься, опираясь руками об колени. Мышечная слабость прогрессирует, у детей развивается сколиоз и сгибательные контрактуры – когда ребенок не может полностью разогнуть конечность. Дети часто падают, поэтому велик риск переломов рук или ног. Отдельные мышечные группы могут замещаться жировой или фиброзной тканью, в результате чего появляется псевдогипертрофия мышц, особенно заметная на лодыжках. Если страдает миокард (сердечная мышца), то существует предрасположенность к развитию нарушений ритма и проводимости сердца, а также дилатационной кардимиопатии (состояния, когда камеры сердца увеличены, а стенки истончены), приводящей к сердечной недостаточности.

В 20-30% случаев при мышечной дистрофии Дюшенна появляются нарушения интеллекта и памяти.
К 12 годам большинство детей вынуждено пользоваться инвалидной коляской. В возрасте 15-20 лет пациентам уже требуется респираторная поддержка, умирают больные миодистрофией Дюшенна от дыхательных или кардиальных осложнений в возрасте 12-25 лет.
Миодистрофия Беккера дебютирует в 10-20 лет и медленно прогрессирует, способность к самостоятельной ходьбе сохраняется в течение 15-20 лет от начала заболевания. Симптоматика схожа с миодистрофией Дюшенна, слабость распространяется на мышцы бедер, таза, плеч, пациенты ходят на носочках или вразвалку, также наблюдается гипертрофия мышц голеней.
Развитие заболевания очень индивидуально, некоторым пациентам требуется инвалидное кресло к 30 годам, некоторые длительное время обходятся тростью.
У пациентов с миодистрофией Беккера также отмечается поражение сердечной мышцы с развитием сердечной недостаточности.
Первые признаки миодистрофии Ландузи-Дежерина проявляются в основном в возрасте 10-20 лет. Сначала атрофия и мышечная слабость наблюдаются в плечевом поясе с поражением мышц лопаток и плеч, потом распространяются на лицо с характерной асимметричностью. Начальными проявлениями являются затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании заболевания страдают лицевые мышцы, при этом пациент не может крепко зажмурить глаза и сжать губы. Позже мимика становится скудной, а речь неразборчивой. Характерными симптомами являются поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира»), «полированный» лоб. Иногда атрофия распространяется на мышцы ног. Другими клиническими признаками миодистрофии Ландузи-Дежерина могут быть аномалии сосудов сетчатки глаза, отек и отслойка сетчатки, снижение слуха.
Для миодистрофии Эмери-Дрейфуса характерны контрактуры локтевых и голеностопных суставов, возникающие в раннем детстве (укорочение ахилловых сухожилий приводит к тому, что ребенок не может опуститься на пятки), тугоподвижность позвоночника, медленно прогрессирующая слабость лопаточно-плечевых и тазово-перонеальных мышц (мышц бедра и голени), а также выраженная кардиомиопатия с нарушениями ритма и проводимости. Тяжесть заболеваний сердца часто определяет прогноз течения болезни вследствие высокой вероятности внезапной сердечной смерти или развития прогрессирующей сердечной недостаточности.
Миодистрофия Эрба-Рота сопровождается слабостью мышц поясничной области и конечностей. Первые признаки появляются в возрасте 10-20 лет: трудности при беге, быстрой ходьбе, прыжках, характерен симптом Говерса. Со временем начинает меняться осанка, походка, снижается тонус мышц плечевого пояса. С прогрессированием заболевания больной может полностью потерять способность ходить. Тотальная гипотрофия мышц туловища приводит к тому, что у пациента начинают выступать лопатки, талия становится очень тонкой, усиливается поясничный лордоз. Характерен симптом свободных надплечий — при попытке приподнять больного, удерживая его подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними.
Диагностика мышечной дистрофии
Предварительный диагноз врач может установить уже при осмотре, наблюдая за попытками ребенка побежать, прыгнуть, подняться по ступенькам, встать с пола.
Для подтверждения диагноза проводятся:
-
Анализ крови на сывороточную креатинкиназу, АСТ, АЛТ.
Креатинкиназа – фермент, катализирующий реакцию фосфорилирования креатина в процессе мышечного сокращения сердечной и скелетноймускулатуры. Тест используют преимущественно в диагностике инфаркта миокарда и миопатий.

