
Спиноцеребеллярная атаксия
Аутосомно-доминантые церебеллярные атаксии. В настоящее время известно более двадцати форм этого заболевания. Клиническая картина в основном определяется поражением мозжечка, иногда страдают также базальные ядра, ствол мозга, спинной мозг, зрительные нервы, сетчатка и спинномозговые нервы. Заболевание может проявляться только мозжечковыми нарушениями или их сочетанием с симптомами поражения перечисленных структур. Изредка развивается деменция. Первыми симптомами заболевания бывает незаметно появляющаяся неловкость, неустойчивость при быстрой ходьбе и беге. Через несколько лет у больного постепенно развивается развернутый атаксический синдром, отмечается неловкость и нарушение координации в руках, интенционный тремор конечностей, адиадохокинез (неритмичность, замедленность движений), скандировання речь, характерным образом нарушается почерк (макрография, неровность строк). Характерны симптомы вовлечения пирамидного и экстрапирамидного тракта, офтальмоплегия (паралич мышц глаза), атрофия зрительных нервов, пигментная дегенерация сетчатки, нистагм (непроизвольные ритмические судорожные движения глазного яблока), амиотрофии.
Молекулярно-генетической причиной заболеваний является увеличение числа тринуклеотидных САG повторов в кодирующем гене. Число повторов обратно пропорционально возрасту манифестации, и прямо пропорционально скорости развития и тяжести заболевания. Характерен феномен антиципации – утяжеление клинических проявлений заболевания из поколения в поколение в пределах одной родословной (более раннее начало и быстрое прогрессирование заболевания, проявление более тяжелых симптомов у потомков). Феномен антиципации обусловлен нестабильностью повтора и нарастанием его длины при передаче мутантного гена от родителя потомкам. Эффект “отцовской передачи” (манифестация более ранних и более тяжелых случаев болезни у потомков больного отца) – обусловлен преимущественным удлинением мутантного повтора в мужском гаметогенезе, тогда как при передаче гена от матери область повтора обычно остается стабильной.
В Центре Молекулярной Генетики проводится прямая молекулярно-генетическая диагностика наиболее частых форм спиноцеребеллярной атаксии: SCA 1, 2, 3, 6, 7, 8, 12 и 17 типов, которая основана на оценке числа CAG-повторов, локализованных в генах ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN8, PPP2R2B и TBP .
Спиноцеребеллярная атаксия 1 (SCA 1, OMIM 164400).
Заболевание обычно начинается в возрасте от 30 до 40 лет (возможный разброс – от 4 до 74 лет). Основные клинические симптомы – атаксия, офтальмоплегия, пирамидные и экстрапирамидные расстройства.
Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных САG повторов в гене ATXN1, располагающемся на 6-й хромосоме (сегмент 6р23). Длина гена составляет 450000 нуклеотидов. Ген содержит девять экзонов. Транскрипт состоит из 10660 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их меньше 36; при болезни больше 40). Нормальные аллели гена обычно содержат в составе тринуклеотидного участка вставки от одного до трех CAT-триплетов, отсутствующие в мутантном гене. Их наличие рассматривается как важный фактор стабилизации нормальных аллелей при мейозе.
Спиноцеребеллярная атаксия 2 (SCA 2, OMIM 183090).
Заболевание обычно начинается в возрасте от 20 до 40 лет (возможный разброс – от 6 до 67 лет). Основные клинические симптомы – атаксия, замедление саккадических движений глазных яблок (саккады – скачкообразные быстрые содружественные фиксирующие движения глаз, возникают, когда взгляд переводится с одного неподвижного предмета на другой), пирамидные и экстрапирамидные расстройства.
Молекулярно-генетической причиной SCA2 является увеличение числа тринуклеотидных САG повторов в гене ATXN2, располагающемся на 12-й хромосоме (сегмент 12q24). Ген содержит 25 экзонов. Длина гена составляет 130000 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их 14-31; при болезни 35-64). Нормальные аллели гена обычно содержат в составе тринуклеотидного участка отдельные вставки CAА-триплетов, стабилизирующих повтор.
Спиноцеребеллярная атаксия 3 (SCA3, болезнь Мачадо-Джозефа, OMIM 109150)
Заболевание обычно начинается после 25-30 лет (возможный разброс – от 5 до 70 лет). Основные клинические симптомы – атаксия (в первую очередь наблюдается абазия (нарушение походки)) офтальмоплегия, парез взора вверх, фасцикуляции мышц лица и фасцикуляции мышц языка, феномен “выпученных глаз” (широко раскрытые глазные щели с фиксированными глазными яблоками), пирамидные и экстрапирамидные симптомы (паркинсонизм). Молекулярно-генетической причиной SCA3 является увеличение числа тринуклеотидных САG повторов в гене ATXN3, располагающемся на 14 хромосоме (сегмент 14q24.3-q31). Ген содержит 11 экзонов. Длина гена составляет 48200 нуклеотидов. В гене имеется участок тринуклеотидных повторов CAG (в норме их 12-47; при болезни 53-86)
Спиноцеребеллярная атаксия 6 (SCA6, OMIM 183086)
Спиноцеребеллярная атаксия 6 типа – аутосомно-доминантное заболевание, частота которого 3:100000. Первый признак – нарушение походки, которое в конечном итоге приковывает пациента к инвалидному креслу. Обычно SCA6 начинается в более позднем возрасте по сравнению с 1-3 типами. Основные клинические симптомы – мягкая прогрессирующая атаксия, нистагм при фиксировании взгляда, дизартрия, дисфагия. При МРТ обнаруживается изолированная атрофия мозжечка. Молекулярно-генетическая причина SCA6 – небольшаяэкспансия тринуклеотидных CAG-повторов, находящихся в 3’ кодирующей области гена CACNA1A, расположенном на хромосоме 19(сегмент 19p13). В норме количество повторов от 5 до 20, при болезни обнаруживается от 21 до 25. Ген CACNA1A кодирует CACNA1А порообразующую субъединицу кальциевого канала, эксперссирующуюся преимущественно в мозжечке (в гранулярных клетках и клетках Пуркинье). Мутации в этом гене приводят к развитию двух других заболеваний – эпизодической атаксии второго типа и семейной гемиплегической мигрени.
Спиноцеребеллярная атаксия 7 (SCA7, OMIM 164500)
Спиноцеребеллярная атаксия 7 типа (оливопонтоцеребеллярная атрофия 3 типа) – прогрессирующее аутосомно-доминантное нейродегенеративное заболевание, клинически характеризующееся церебеллярной атаксией, ассоциированной с дистрофией желтого пятна. Средний возраст манифестации заболевания – 32 года. Степень тяжести, скорость прогрессии и возраст начала заболевания варьируют как между семьями так и внутри семей. Основные клинические симптомы – офтальмоплегия, пирамидные и экстрапирамидные знаки, дизартрия, дисфагия, хорея, гиперрефлексия, спастика, потеря глубокой чувствительности, пигментная дегенерация сетчатки, прогрессирующая потеря зрения, медленные саккады, атрофия зрительного нерва.
Молекулярно-генетическая причина SCA7 –экспансия тринуклеотидных CAG-повторов гена ATXN7 (3p21.1-p12), находящихся в полиглутаминовом тракте белка ataxin-7.В норме количество повторов варьирует от 4 до 35, при болезни обнаруживается от 36 до 306 повторов.
Спиноцеребеллярная атаксия 8 (SCA8, OMIM 608768)
Спиноцеребеллярная атаксия 8 типа – медленно прогрессирующее аутосомно-доминантное заболевание, возраст начала которого варьирует от 18 до 65 лет. Основные клинические симптомы – прогрессирующая церебеллярная атаксия, нарушение координации походки, движения конечностей, речи, брадикинезия (замедленные движения). У больных часто наблюдается дизартрия, тремор, дисфагия, нистагм, замедление саккадических движений глазных яблок, дизметрические саккады, потеря чувствительности. При МРТ обнаруживается атрофия полушарий и червя мозжечка. Молекулярно-генетической причиной SCA8 является увеличение числа тринуклеотидных СAG повторов в гене ATXN8. В норме количество повторов варьирует от 15 до 50, при болезни обнаруживается от 71 до 1300.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий – около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Спиноцеребеллярная атаксия 12 (SCA12, OMIM 604326)
Спиноцеребеллярная атаксия 12 типа – аутосомно-доминантное заболевание. Начинается в возрасте от 8 до 55 лет. Основные клинические симптомы – тремор верхних конечностей, тремор головы, атаксия, дисметрия, дисдиадохокинез, гиперрефлексия, скудость движения, аномальные движения глаз, деменция. При МРТ и КТ обнаруживается атрофия коры головного мозга и мозжечка.
Молекулярно-генетическая причина SCA12 – увеличение числа тринуклеотидных CAG повторов в гене PPP2R2B, распологающемся на 5-ой хромосоме (сегмент q31-q33). В норме количество повторов от 7 до 32, при болезни обнаруживаются от 51 до 78. Клиническое значение экспансии повторов в диапазоне от 33 до 50 до сих пор не установлено. Ген PPP2R2B кодирует регуляторную субъединицу В белка фосфатазы 2 участвующего в таких регуляторных процессах, как рост и деление клеток, сокращение мышц и транскрипция генов.
Спиноцеребеллярная атаксия 17 (SCA 17, OMIM: 607136)
Спиноцеребеллярная атаксия 17 типа – аутосомно-доминантное неврологическое расстройство, характеризующееся атаксией, пирамидными и экстрапирамидными расстройствами (паркинсонизм), когнитивными нарушениями, психозом и судорогами. Клинические проявления схожи с Хореей Гентингтона. Заболевание обычно начинается в возрасте от 23 до 29 лет (возможный разброс – от 3 до 53 лет).
Молекулярно-генетической причиной SCA17 является увеличение числа тринуклеотидных CAG (или CAA) повторов в гене TBP, расположенном на длинном плече 6 хромосомы (сегмент q27). Ген TBP кодирует фактор транскрипции-белок, связывающий последовательность ТАТА-бокса (ТВР). В норме количество повторов варьирует от 25 до 44, при болезни обнаруживается от 47 до 63 повторов. Описана связь 45 и 46 повторов с неполной пенетрантностью заболевания.
Спиноцеребеллярная атаксия
Нейродегенерация – патологический процесс, с развитием которого нервная ткань утрачивает свою сложнейшую организацию, вырождается, постепенно атрофируется (уменьшается в объеме и отмирает), становясь, в целом, функционально несостоятельной. Учитывая, что нервная система контролирует и регулирует в организме буквально всё, нейродегенеративные заболевания, – даже самые медленные и вялотекущие, – всегда составляют серьезную проблему, которая усугубляется еще и тем, что на данный момент все усилия по разработке репаративных (восстановительных) и этиопатогенетических (устраняющих первопричину болезни) видов терапии не принесли ощутимых результатов.
В большинстве своем нейродегенеративные болезни обусловлены или, по крайней мере, достоверно связаны с наследственными, хромосомными факторами. Эти заболевания традиционно считаются редкими, и в пересчете на десятки и сотни тысяч населения многие из них действительно кажутся спорадическими, почти случайными аномалиями. Однако если просуммировать частоту встречаемости достаточно известных болезней Альцгеймера, Пика, Паркинсона, демиелинизирующего рассеянного или бокового амиотрофического склероза, ДТЛ (деменция с тельцами Леви), картина будет выглядеть более тревожной. Так, со ссылкой на данные посмертных патоморфологических исследований в литературе неоднократно подчеркивалось, что та же ДТЛ (один из лобно-височных вариантов нейродегенерации) диагностируется значительно реже, чем в действительности встречается.
Большое число отдельных нозологических единиц (т.е. официально устанавливаемых диагнозов этой группы) служит предметом критических дискуссий, поскольку дегенерация нейронной ткани является основным и общим механизмом развития таких болезней; напр., крайнее крыло сторонников обобщения предлагало «для удобства» считать все заболевания такого рода лишь частными вариантами болезней Альцгеймера или Паркинсона. Едва ли такой подход можно считать оправданным: клиническая картина, темпы протекания, прогноз, стратегия симптоматического лечения – все это детерминируется рядом значимых факторов (прежде всего, преимущественной локализацией процесса) и отличается в достаточной степени, чтобы говорить именно о самостоятельных заболеваниях.
Вышесказанное в полной мере относится к спиноцеребеллярной атаксии. Это наследственное нейродегенеративное заболевание с выраженной собственной спецификой, которое может манифестировать в любом возрасте (обычно в интервале 5-40 лет) и отличается многообразием форм: к настоящему времени выделено и описано свыше двадцати сравнительно самостоятельных типов (SCA, SCA 2, болезнь Фридрейха и мн.др.).
2. Причины
В основе спиноцеребеллярных атаксий лежит наследуемая мутация определенных генов (тип наследования, как правило, таков, что если оба родителя являются носителями, то вероятность «срабатывания» патологии у ребенка составляет 1/4 или 25%). В результате нарушается ряд сложнейших электрохимических процессов, управляющих энерго- и белковым балансом (значительную роль играет дефектная структура белка фратаксина), передачей нервных импульсов от центра к периферии и обратно, и пр. Объединяющей особенностью для всей группы атаксий является то, что в нейронную дегенерацию вовлекаются структуры как головного мозга (прежде всего, мозжечок), так и спинного, а также проводниковые пути между ними, периферические нервы и, в отдельных случаях, ткань миокарда. Термин «атаксия» в дословном переводе означает «отсутствие порядка, согласованности», и, по определению, главным проявлением спиноцеребеллярной атаксии становятся прогрессирующие нарушения координации движений и, вообще, нервно-мышечной согласованности.
3. Симптоматика, диагностика
Вероятные симптомы спиноцеребеллярной нейронной дегенерации настолько полиморфны, что описать хотя бы основные из двадцати ее типов в одной статье нет никакой возможности. Отмечаются расстройства зрительно-моторной координации и мышечного тонуса (тремор, экстрапирамидная «скованность»); частичные параличи глазодвигательных мышц и дегенеративные ретинопатии в сочетании с атрофией зрительного нерва; общая атрофия мышечных волокон; разнообразные неврологические симптомокомплексы. Если нейродегенеративный процесс затрагивает кору головного мозга, постепенно развивается деменция – ослабоумливающее снижение когнитивных функций (память, внимание, различные виды распознавания и пр.), логического мышления, организации речи. В случае поражения продолговатого мозга развивается «бульбарная» симптоматика: деградация глотательного, небного, жевательного, дыхательного рефлексов.
Заболевания этой группы прогрессируют относительно медленно: течение может занимать до 20 лет и более, хотя описаны и значительно более быстрые развития. Больные постепенно утрачивают способность к самообслуживанию и, вообще, к продуктивному контакту с миром; они все больше зависят от опеки и ухода со стороны окружающих, к терминальной стадии впадая в полную беспомощность и погибая, как правило, от присоединившихся пневмоний, истощения, дыхательной недостаточности и пр.
Нейродегенеративные заболевания, в том числе атаксии, диагностируются клинически, в ходе неврологического осмотра и тщательного анализа жалоб и анамнестических сведений. Дополнительно могут назначаться методы томографической визуализации, нейропсихологическое обследование и пр., однако окончательно диагноз устанавливается и дифференцируется, как правило, лишь патоморфологически.
4. Лечение
В настоящее время не существует лечения, которое обращало бы вспять или хотя бы останавливало процессы нейродегенерации. Все виды практикуемой сегодня терапии носят сугубо паллиативный характер и направляются на смягчение наиболее дезадаптирующих, снижающих качество жизни симптомов, доминирующих в конкретной клинической картине. Как правило, назначают препараты для улучшения нейротрофики (питания нервных тканей), витаминные комплексы, массаж, лечебная физкультура для коррекции и/или компенсации двигательных расстройств.
Спиноцеребеллярная атаксия: причины, симптомы, методы диагностики и лечения
Заболевания нервной системы – не редкость. Они чаще всего проявляются невралгиями, но существуют и наследственные формы болезней, например, спиноцеребеллярная атаксия (СА). Неврологи используют термин «атаксия» для обозначения состояния, при котором наблюдается нарушение слаженности движений, контролирующихся сознанием. Иными словами, данный недуг представляет собой нарушение совместной деятельности мозжечка и спинного мозга. В чем проявляется недуг и как он лечится, рассмотрим ниже.
Описание
Атаксия спиноцеребеллярная представляет собой совокупность генетических недугов, которые носят неврологический характер, проявляются нарушением деятельности базальных ядер головного мозга и мозжечка и передаются по наследству. В результате этого изменяется координация движений и прочее. Негативным в заболевании является тот факт, что на сегодняшний день какого-либо определенного лечения не существует. Болезнь наследуется по механизму аутосомной доминантности, когда ферменты, которые появляются в результате мутаций генов структурных белков, деформируются. Обычно наследование происходит по отцовской линии из поколения в поколение.
Этиология
Поскольку спиноцеребеллярная атаксия является болезнью наследственной, то она встречается в каждом последующем поколении, где был больной отец. Заболевание само по себе не слишком распространено (от одного до двадцати четырех заболевших на сто тысяч человек). При этом разные типы недуга встречаются в разных странах мира. В современной медицине существует больше двадцати вариантов этой болезни.
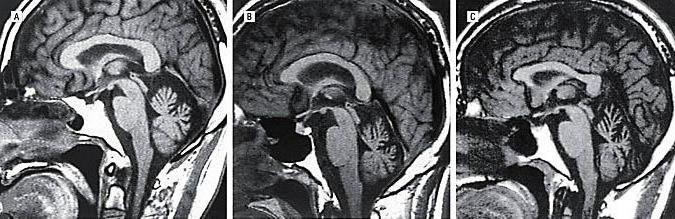
Классификация
Спиноцеребеллярная атаксия (мкб 10 – G11) в 90% случаев представляет собой шесть из двадцати генетических вариантов болезни. Данные варианты были классифицированы по номерам: 1, 2, 3, 6, 7 и 8 типы атаксии. Патология выражается в изменении количества CAG в части генов, которые кодируют больные гены. Рассмотрим далее эти типы подробнее:
- Атаксия первого типа сегодня самая распространенная. Она возникает вследствие размещения в шестой хромосоме мутированного гена ATXN1. В норме этот ген имеет тридцать шесть повторов, при большем же их количестве развивается болезнь. Мутация гена вызывает образование ДНК белка, который принимает участие в метаболизме клеток. Это способствует дегенерации и развитию болезни.
- СА второго типа распространена несколько меньше. Она характеризуется увеличением повторов в двенадцатой хромосоме. Какую функцию при этом выполняет белок, медицине неизвестно.
- Третий тип именуется болезнью Мачадо-Джозефа. В этом случае нарушение происходит в гене, что размещен в четырнадцатой хромосоме. Белок при этом принимает участие в обмене энергией между мозжечком и ядрами мозга.
- Спиноцеребеллярная атаксия шестого типа является редким недугом. Здесь происходит нарушение в гене, который находится в девятнадцатой хромосоме. Ген кодирует белок, который размещается в нейронах мозжечка. Этот процесс вызывает также наследственную форму мигрени.
- СА седьмого типа обуславливается нарушениями гена в третьей хромосоме. Какую функцию выполняет белок, медицине неизвестно.
- Восьмой тип характеризуется изменениями гена в тринадцатой хромосоме.
Причины

При любом типе заболевания происходит мутация гена, приводящая к образованию ДНК белка патологической формы, который богат глутамином. Он вызывает появление в ядрах нейронов мозжечка и базальных ядер мозга отложений в виде агрегатов, нарушая свойства протеинов. Белки принимают участие в обмене веществ, протекающих в нервной ткани. Скорость протекания данного процесса зависит от количества поворотов в гене, которое отличается от нормы. Это определяет симптоматику заболевания. При созревании половых клеток симптомы усиливаются.
Симптомы
Все типы данного заболевания имеют одинаковую симптоматику, различными могут быть только второстепенные элементы. Так, спиноцеребеллярной атаксии симптомы не проявляются в детском возрасте. Средний возраст людей, страдающих недугом, составляет от восемнадцати до тридцати лет. Атаксия третьего, шестого и седьмого типов развивается позже, обычно это происходит после тридцати лет. Первым признаком недуга является появление неуклюжести при ходьбе и беге. Позже наблюдается тремор конечностей, изменение походки, офтальмоплегия, почерк меняется. Со временем недуг приводит к развитию паркинсонизма. При некоторых видах болезни наблюдается атрофия зрительного нерва. СА шестого, седьмого и восьмого типов характеризуется нарушением речи и процесса глотания, что приводит к истощению. Истощение вместе с патологиями часто провоцируют смертельный исход. При всех видах заболевания нарушается координация движений. Средняя продолжительность жизни больных составляет от десяти до двадцати пяти лет, в зависимости от формы недуга и качества ухода за ними.

Диагностика
Прежде всего, проводят неврологический осмотр больного, изучают анамнез, проводят МРТ и различные молекулярно-генетические анализы и ДНК -диагностики . На разных стадиях развития недуга врач выявляет разные нарушения, связанные с неврологией. Это может быть тремор, нарушение речи, дисфагия и прочее. Некоторые формы заболевания обуславливаются быстрым развитием зрительных нарушений, что приводят к полной слепоте. Заболевание склонно прогрессировать. При исследовании наследственного анамнеза может быть обнаружено аутосомно-доминантное наследование от отца. МРТ показывает нарушения в области больших полушарий, мозжечка и базальных ядер. Может также наблюдаться атрофия мозжечка. Молекулярное исследование и ДНК- диагностика указывают на увеличенное число повторов в генах больного.
Лечение
Какого-либо эффективного лечения данного недуга на сегодняшний день нет. Результативность поддерживающей терапии на данный момент не доказана, однако она проводится для замедления развития недуга. Так, спиноцеребеллярная атаксия лечение предполагает в виде витаминотерапии и средств, которые стимулируют обмен веществ и метаболизм в нервной ткани. Также больным назначают ноотропные препараты. Немаловажную роль играет и физическая культура. Врачи рекомендуют больным выполнять комплекс упражнений для укрепления мышц и уменьшения нарушений равновесия. Проводят сеансы массажа и электромиостимуляцию.
Прогноз
Как правило, прогноз при данном заболевании неблагоприятный, поскольку недуг постоянно прогрессирует и приводит к инвалидизации, а затем и к летальному исходу. Поэтому ответ на вопрос о том, лечится ли спиноцеребеллярная атаксия, будет отрицательным. Данное наследственное заболевание неизлечимо. В некоторых случаях прогноз может быть не столь негативным. Это бывает при развитии заболевания в преклонном возрасте и своевременном лечении, тогда большое количество симптомов могут не проявиться. Если недуг обнаружился в молодом возрасте, длительность жизни таких пациентов будет невелика. Больные с пятым и шестым типом недуга живут нормальной жизнью немного дольше, обычно срок их жизни не меняется. При правильном уходе и своевременной терапии врачам удается увеличить время жизни больных на десять лет. В среднем с таким заболеванием, как спиноцеребеллярная атаксия, живут около двадцати лет. Причиной летального исхода часто становится сердечная недостаточность и наличие инфекций.
Профилактика
Профилактические меры представляют собой медицинское и генетическое консультирование родителей, в чьем анамнезе наблюдались такие состояния. Также проводится генетическая перинатальная диагностика. Врачом определяется риск появления недуга у прямых родственников. Риск развития патологии для здоровых братьев и сестер, а также детей больного составляет 50%. В свою очередь, дети этих людей имеют вероятность унаследовать болезнь в 25%. Все эти лица находятся в группе риска и представляют собой главные объекты для консультирования. Основой профилактического исследования является ДНК-диагностика лиц из группы риска на наличие мутированных генов.
Спиноцеребеллярная атаксия в современное время является тем заболеванием, которое не лечится и приводит со временем к летальному исходу. Предупредить развитие заболевания можно при помощи специальных методов диагностики, но предотвратить его развитие невозможно, поскольку недуг этот имеет наследственный характер и обуславливается мутациями здоровых генов. Все это подталкивает современную медицину к разработке методов исследования на самых ранних этапах развития болезни, а также изучению причин мутаций, которые передаются по наследству от отца к детям.
Спиноцеребеллярные атаксии
Спиноцеребеллярные атаксии – группа генетически разнородных наследственных заболеваний неврологического характера, которые проявляются различными расстройствами работы мозжечка и иногда базальных ядер головного мозга. Симптомами этого состояния являются: развитие атаксии и неустойчивой походки, нарушение координации движений и другие неврологические проявления. Диагностика спиноцеребеллярных атаксий производится на основании данных неврологического осмотра, изучения наследственного анамнеза больного, магнитно-резонансной томографии и молекулярно-генетических исследований. Специфического лечения этой патологии на сегодняшний момент не существует, для сохранения оптимального качества жизни больного используют методы поддерживающей и симптоматической терапии.
Общие сведения
Спиноцеребеллярные атаксии – группа наследственных неврологических состояний, характеризующихся развитием прогрессирующей дегенерации клеток мозжечка и иногда базальных ядер вплоть до их полной атрофии. Впервые одно из заболеваний этой группы было описано еще в 1891 году немецким невропатологом П. Менцелем, который выявил развитие атаксии, офтальмоплегии и других неврологических нарушений в рамках одной семьи. Дальнейшие исследования показали, что это состояние (известное сейчас как спиноцеребеллярная атаксия 1-го типа) наследуется по аутосомно-доминантному механизму.
В настоящий момент методами современной генетики удалось обнаружить более 20 различных генетических вариантов этого заболевания, при этом более 90% всех случаев обуславливает только 6 из них (1, 2, 3, 6, 7 и 8-й типы). Все формы спиноцеребеллярных атаксий характеризуются аутосомно-доминантным наследованием с явлениями антиципации (усиления выраженности патологии от поколения к поколению) и «отцовской передачи» – более яркой клинической картине заболевания при его наследовании от отца. Поэтому в ряде регионов в общей структуре патологии наблюдается незначительное превалирование больных мужского пола. Общая встречаемость спиноцеребеллярной атаксии колеблется в широких пределах (1-24:100 000), при этом 1-й тип распространен в России и большей части Европы, 2-й – в Индии, 3-й – в Германии и Японии.
Причины и классификация спиноцеребеллярных атаксий
Несмотря на значительное генетическое и отчасти клиническое разнообразие спиноцеребеллярных атаксий, молекулярные механизмы генетических нарушений при этих заболеваниях очень сходны. Основная причина патологии заключается в изменении количества тринуклеотидных последовательностей (CAG) в кодирующей части ассоциированных с заболеванием генов. Это приводит к увеличению количества аминокислоты глутамина в полученном белке, что изменяет физико-химические свойства протеина и нарушает его функции. В ряде случаев вышеуказанные белки прямо или косвенно участвуют в метаболизме нервной ткани, поэтому изменение их структуры приводит к спиноцеребеллярной атаксии. В настоящее время лучше всего изучены молекулярные механизмы 6 основных разновидностей этого заболевания – данные формы патологии встречаются наиболее часто и в совокупности составляют более 90% случаев спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 1-го типа считается самым распространенным и самым изученным вариантом данной патологии. Ее причиной выступают мутации в гене ATXN1, который располагается на 6-й хромосоме. В норме данный ген имеет не более 36 CAG-повторов, увеличение их количества приводит к развитию заболевания. Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий белок, активно участвующий в метаболизме клеток Пуркинье мозжечка – при наличии мутантной разновидности гена это приводит к появлению агрегантов и постепенной дегенерации, что и становится причиной спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 2-го типа – менее распространенный вариант заболевания, этиология не так тщательно изучена. Причиной патологии является увеличение количества CAG-повторов в гене ATXN2, локализованном на 12-й хромосоме. В здоровом варианте гена количество вышеуказанных последовательностей составляет от 15 до 36, тогда как при спиноцеребеллярной атаксии их может быть свыше 100. Функции белка, который кодируется геном ATXN2, на сегодняшний момент неизвестны.
Спиноцеребеллярная атаксия тип 3 (другое название – болезнь Мачадо-Джозефа в честь двух больных, у которых впервые было описано данное состояние) – причиной этого варианта патологии выступают нарушения в гене ATXN3, расположенном на 14-й хромосоме. В норме количество CAG-повторов в этом гене не превышает 47, при развитии заболевания обнаруживается от 53 до 68 повторов. Данный ген кодирует белок, который предположительно участвует в энергетическом обмене нейронов мозжечка и базальных ядер.
Спиноцеребеллярная атаксия тип 6 – сравнительно редкий вид заболевания, обусловленный дефектами в гене CACNA1A, локализованном на 19-й хромосоме. Для развития патологии достаточно очень незначительного увеличения количества CAG-повторов – если в нормальном варианте гена их обнаруживают 5-20, то при наличии атаксии – 21-26. Ген CACNA1A кодирует белок-субъединицу кальциевых каналов, расположенных на нейронах мозжечка. Помимо спиноцеребеллярной атаксии, нарушения в гене CACNA1A обуславливают развитие эпизодической атаксии и некоторые наследственные формы мигрени.
Спиноцеребеллярная атаксия тип 7 – данная разновидность патологии вызывается нарушениями структуры гена ATXN7, который располагается на 3-й хромосоме. У здорового человека количество CAG-повторов составляет не более 35, тогда как при заболевании их количество может достигать нескольких сотен. Функции белка, который кодирует ген ATXN7, на сегодняшний момент изучаются.
Спиноцеребеллярная атаксия тип 8 обусловлена генетическим дефектом гена ATXN8, расположенного на 13-й хромосоме. Как и в других случаях, суть генетического дефекта при этом состоянии заключается в изменении количества тринуклеотидных последовательностей CAG – обычно их около 15-50, тогда как при патологии количество повторов может составлять свыше 1200.
Практически при любом типе спиноцеребеллярной атаксии патологическая форма белка, чрезмерно богатая глутамином, формирует отложения в ядрах или цитоплазме нейронов мозжечка и базальных ядер в виде плотных агрегатов. Этот процесс идет тем быстрее, чем сильнее количество CAG-повторов в ключевом гене отличается от нормы. Этим же объясняется механизм антиципации симптомов спиноцеребеллярной атаксии – в процессе мейоза при образовании половых клеток количество вышеуказанных тринуклеотидных последовательностей может увеличиваться, что приводит к усилению симптомов.
Так как подобное явление чаще имеет место при формировании мужских половых клеток, это становится причиной так называемой «отцовской передачи», когда антиципация регистрируется только при передаче заболевания от отца потомству. Многие врачи-генетики полагают, что основная причина спиноцеребеллярных атаксий лежит не в увеличении «гистидиновых» тринуклеотидов, а в делеции так называемых регулирующих триплетов, разделяющих участки CAG-повторов. Например, при первом типе заболевания это CAT, при втором CAA – они регулируют количество CAG-повторов и сохраняют стабильность их количества во время мейоза.
Симптомы спиноцеребеллярных атаксий
Несмотря на значительное генетическое разнообразие спиноцеребеллярных атаксий, проявления разных типов этого заболевания в целом сходны и различаются только второстепенными деталями – возрастом манифестации, особенностями некоторых симптомов. Практически все формы патологии не регистрируются в детском возрасте – лишь отдельные случаи 1 и 2-го типов были замечены у детей младше 7 лет, средний возраст их манифестации – 18-30 лет. Спиноцеребеллярные атаксии 3, 6 и 7-го типов характеризуются еще более поздним развитием – их манифестация практически всегда происходит у лиц старше 30 лет. Нередко подобные нарушения выявляются и у пожилых людей, что затрудняет дифференциальную диагностику этого состояния с болезнью Паркинсона и другими нейродегенеративными заболеваниями старшего возраста.
Чаще всего развитие спиноцеребеллярной атаксии начинается с появления простой неуклюжести в движениях, особенно при ходьбе, беге. В дальнейшем возникает тремор рук, нарушения походки, паралич глазодвигательных мышц (офтальмоплегия), изменяется почерк больного (становится крупнее, строки неровные). В конечном итоге заболевание приводит к выраженной мозжечковой атаксии, расстройствам пирамидальных и экстрапирамидальных путей, паркинсонизму. Некоторые формы патологии характеризуются выраженными нарушениями зрения – развитием атрофии зрительного нерва, пигментной дегенерации сетчатки и других процессов.
Спиноцеребеллярная атаксия 6, 7 и 8-го типов также проявляется расстройствами речи (дизартрия) и глотания, что является причиной затрудненного питания и истощения больных. Именно это обстоятельство и связанные с ними нарушения (например, атрофия мозжечка, сердечная недостаточность) часто становятся причиной смерти пациентов. В зависимости от формы заболевания, объема поддерживающего лечения и качества ухода за больными продолжительность жизни при спиноцеребеллярной атаксии может составлять от 10 до 25 лет с момента возникновения первых симптомов патологии.
Диагностика
Выявление спиноцеребеллярной атаксии производится на основании данных неврологического осмотра, изучения наследственного анамнеза, магнитно-резонансной томографии головного мозга и молекулярно-генетических исследований. При осмотре больных на разных стадиях развития патологии определяются различные по выраженности неврологические нарушения – тремор конечностей, атаксия, изменения речи и голоса, на конечных этапах – дисфагия. Некоторые формы спиноцеребеллярной атаксии сопровождаются достаточно быстрым развитием нарушений зрения, приводящим к полной слепоте. Многолетнее наблюдение за такими больными подтверждает неуклонно прогрессирующее течение заболевания. При изучении наследственного анамнеза могут определяться характерные признаки спиноцеребеллярной атаксии – аутосомно-доминантное наследование, наличие антиципации при передаче болезни от отца.
На МРТ головного мозга при спиноцеребеллярной атаксии обнаруживаются очаги демиелинизации и нейродегенерации в области полушарий, червя мозжечка и базальных ядер. На терминальных стадиях развития заболевания может отмечаться полная атрофия мозжечка. Молекулярно-генетические исследования при спиноцеребеллярной атаксии сводятся к поиску патологически увеличенного количества CAG-повторов в генах, ассоциированных с этим заболеванием. В настоящее время большинство лабораторий мира осуществляет поиск этого дефекта в генах, наиболее часто приводящих к развитию патологии – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 и CACNA1A.
Лечение спиноцеребеллярной атаксии
Специфическое лечение патологии отсутствует, поддерживающая терапия способна несколько замедлить развитие спиноцеребеллярной атаксии, но единого мнения по поводу ее эффективности на сегодняшний момент нет. Применяют витаминотерапию (Е, А, группы В), ноотропные средства, стимуляторы обмена веществ (рибоксин) и метаболизма в нервной ткани. При развитии непроизвольных движений рекомендуют использовать клоназепам и галоперидол. Важную роль в сдерживании прогрессирования спиноцеребеллярной атаксии играет лечебная физкультура – регулярное выполнение правильно подобранного комплекса упражнений позволяет укрепить мышцы и снизить выраженность расстройств равновесия. С этой же целью рекомендуют проведение сеансов лечебного массажа, процедуры электромиостимуляции.
Прогноз и профилактика
В долгосрочной перспективе прогноз любой формы спиноцеребеллярной атаксии неблагоприятный – это заболевание характеризуется выраженным прогрессирующим течением и со временем приводит сначала к инвалидизации, а затем к смерти больного. Однако в конкретном случае прогноз может быть и менее негативным – например, при развитии патологии в пожилом возрасте и своевременно начатом поддерживающем лечении большинство тяжелых симптомов попросту не успеет проявиться. Если спиноцеребеллярная атаксия возникла в молодом или детском возрасте, продолжительность жизни таких больных даже при интенсивном лечении и тщательном уходе будет резко снижена.
Профилактика осуществляется методом медико-генетического консультирования родителей, наследственный анамнез которых отягощен по этому состоянию, и генетической пренатальной диагностики. При этом необходимо учитывать аутосомно-доминантный характер наследования спиноцеребеллярной атаксии и такие особенности ее передачи, как антиципация.
Виды спиноцеребеллярной атаксии
Что такое спиноцеребеллярная атаксия?
Атаксия — дегенеративное заболевание, влияющее на нервную систему, вызывающую плохую координацию, нарушения движений, трудности с речью, ходьбой, двигательными навыками, глотанием и зрением. В основном это затрагивает взрослых людей.
Спиноцеребеллярная атаксия (SCA) представляет собой форму наследственной прогрессирующей атаксии с более чем 20 идентифицированными типами, которые имеют похожие симптомы. В настоящее время нет какого то конкретного лечения этого заболевания или лечения способствующего замедлению прогрессирования спиноцеребеллярной атаксии.
Причины атаксии:
Повреждение мозжечка (части мозга, которая отвечает за координацию движения) обычно является причиной атаксии. Однако повреждение других участков нервной системы может также вызвать атаксию. Эти нарушения могут быть вызваны травмой или кислородным голоданием головного мозга, чрезмерным потреблением алкоголя в течение длительного периода времени или существующим состоянием, таким как к примеру рассеянный склероз (MS). Атаксия также может быть наследственной, вызванной нарушением генотипа, передаваемым членами семьи, у которых не всегда появляются признаки атаксии.
Существуют три основные категории атаксии:
Приобретенная (негенетическая) атаксия
Приобретенная атаксия обычно вызвана внешними факторами, такими как травмы, опухоль или химическое воздействие, и может воздейстовавать на все возрастные группы. Она быстро развивается, в течение нескольких часов или дней. Она может как ухудшаться, так и улучшаться со временем или оставаться неизменной.
Наследственная атаксия
Атаксия Фридриха является наиболее распространенной формой наследственной атаксии, затрагивающей около 50 000 человек с симптомами, начинающимися в детском или подростковом возрасте. Люди с атаксией Фридриха обычно имеют более короткую продолжительность жизни, чем обычно, поскольку это оказывает разрушительное воздействие в том числе и на сердце.
Идиопатическая поздняя мозжечковая атаксия (ILOCA)
ILOCA начинается примерно в возрасте 50 лет и старше и её симптомы со временем ухудшаются.
Спиноцеребеллярная атаксия
Спиноцеребеллярная атаксия является наследственной формой атаксии, поражающей людей в возрасте от 25 до 80 лет, и характеризуется:
• Проблемами с балансом и координацией
• Дизартрией (невнятная и медленная речь)
• Дисфагей (затруднение при глотании)
• Судорогами и мышечной напряженностью жесткость
• Периферическая невропатия (потеря чувства в руках и ногах)
• Потерей памяти
• Медленным движением глаз
• Недержанием мочи
В зависимости от типа SCA могут присутствовать различные симптомы.
Причины спиноцеребеллярной атаксии
Причиной спиноцеребеллярной атаксии является атрофия мозжечка, как видно из других форм атаксии. Начало спиноцеребеллярной атаксии обычно происходит после 18 лет и прогрессирует медленно, при этом симптомы ухудшаются в течение нескольких лет. Некоторые типы SCA могут развиваться быстрее.
SCA можно унаследовать аутосомно-доминантным способом. Симптомы могут присутствовать только от одной мутированной копии ответственного гена в каждой клетке, но некоторые случаи вызваны разрастанием тринуклеотидных повторов, где фрагмент ДНК повторяется много раз. Эти повторы не всегда вызывают проблемы. Часто, чем больше количество повторов, тем раньше это происходит и хуже симптомы.
Симптомы спиноцеребеллярной атаксии
Клинические особенности, отличные от других форм атаксии, включают несколько не мозжечковых признаков, в том числе:
• паркинсонизм
• хорея (непроизвольное нарушение движения)
• когнитивные нарушения
• периферическая невропатия
• приступы
Диагностика
Атаксия диагностируется с помощью сбора анамнеза, изучения семейной истории, клинико-лабораторными методами (анализы крови, чтобы исключить другие состояния, представляющие подобные симптомы, и неврологические оценки). 12 типов атаксии, атаксия Фридриха и ряд других атаксией потребуют генетических анализов крови для диагностики; однако некоторые формы SCA не могут быть точно диагностированы, поскольку они не были генетически идентифицированы ранее. В спиноцеребеллярной атаксии это составляет примерно 25-40% случаев. Поскольку существует совпадение симптомов между различными типами атаксии спиноцеребелляров, генетическое тестирование может быть использовано для конкретной диагностики типа SCA, влияющего на пациента. В этих случаях для диагностики SCA могут использоваться неврологические исследования, которые могут включать МРТ-исследование головного мозга и позвоночника.
Лечение
В настоящее время нет какого то конкретного лечения этого заболевания или лечения способствующего замедлению прогрессирования спиноцеребеллярной атаксии. Пациентам приходится тесно общаться с неврологом, чтобы разработать персональный план борьбы с симптомами атаксии, который может включать речевую терапию, профессиональную терапию и физическую терапию. Лекарства можно назначать в сочетании с терапией, чтобы помочь справиться с симптомами.
Прогноз спиноцеребеллярной атаксии
Прогноз для людей с спиноцеребеллярной атаксией является переменным и отличается в зависимости от типа SCA. Прогноз часто основывается на наиболее распространенных типах SCA, SCA1, SCA2, SCA3 и SCA6. Те, у кого эти типы SCA, как правило, нуждаются в инвалидной коляске через 10-15 лет после появления симптомов, и помощь в ежедневных задачах будет необходима.
В стоимость МРТ исследования входит заключение врача-рентгенолога, диск со всеми снимками и выборочные снимки на фотобумаге.
В стоимость КТ исследований входит заключение врача-рентгенолога и диск со всеми снимками.
При необходимости печати снимков на рентгеновской пленке – доплата составит 300 рублей.

Заказать звонок
Имеются противопоказания, необходима консультация врача.
Лицензия на осуществление деятельности ЛО-66-01-006579 от 23.07.2020
ООО «СканЛайн», юридический адрес: 620049, г. Екатеринбург, пер. Автоматики, стр. 1, комн. 51
О правах и обязанностях граждан в сфере охраны здоровья
Спиноцеребеллярная атаксия: признаки, способы диагностики и лечения
Спиноцеребеллярная атаксия 1 типа
Спиноцеребеллярная атаксия 1 типа (SCA 1)- тяжелое нейродегенеративное прогрессирующее заболевание с поздним возрастом манифестации, наследуется по аутосомно-доминантному типу; клинически характеризуется сочетанием нарастающих расстройств координации движений с признаками мультисистемного поражения головного и спинного мозга (Иллариошкин и др., 2002). Начало симптомов при СЦА1 – наблюдается обычно во взрослой жизни, в среднем – в 30 лет. Если симптомы появляются раньше – до 20 лет, дополнительно к атаксии часто встречаются и другие симптомы. В случаях очень раннего (до 13 лет) болезнь может быть более тяжелой и иметь быстрое прогрессирование.
 |
Основные клинические симптомы: первым симптомом обычно является нарушение координации рук и нарушение баланса при ходьбе. Фактически, само слово “атаксия” означает нарушение координации. При прогрессировании СЦА1 в течение нескольких лет появляются трудности при глотании и неясная речь. В некоторых случаях, у больных появляются дополнительные признаки типа нейропатии (потери чувств и рефлексов в ногах), мышечной спастичности, слабости, или потери памяти. |
При СЦА1 генетические дефекты ведут к ухудшению работы определенных нервных волокон, несущих информацию к головному мозгу и от него, которая заканчивается дегенерацией мозжечка (координационного центра мозга). Часто отмечается кровное родство родителей. Заболевание не поддается лечению, смерть чаще всего наступает спустя 10-15 лет от момента появления первых симптомов. Генетическая причина развития СЦА1 заключается в экспансии числа копий тандемных тринуклеотидных CAG-повторов в кодирующей области гена SCA1 (Orr et al., 1993). Для СЦА1 возможна прямая ДНК-диагностика заболевания на пресимптомной стадии, иногда за много лет до появления каких-либо неврологических и/или психических расстройств. Это имеет особое значение для медико-генетического консультирования, поскольку в настоящее время не найдены еще эффективные способы лечения СЦА1, хотя исследования в этом направлении активно ведутся в мире (Ogawa, 2004; Dueñas et al., 2006; Takei et al., 2007; Gao et al., 2008). Единственным способом борьбы с этим заболеванием на сегодняшний день является профилактика появления новых случаев СЦА1 в отягощенных семьях (Иллариошкин и др., 1997). До середины 70-х годов в Якутии болезнь относили к “мозжечковой форме” вилюйского энцефаломиелита. В настоящее время спиноцеребеллярная атаксия I типа рассматривается как этноспецифическое наследственное заболевание (Пузырев, Максимова, 2008).
Высокая распространенность заболевания (38,6 на 100 тыс. якутов по сравнению 1-2:100 тыс. в мировом населении) в Якутии была оценена как “сибирский очаг” накопления заболевания, крупнейший в мире. Районы высокого накопления СЦА1 – Абыйский и Усть-Алданский улусы Якутии – характеризуются однородным национальным составом; высоким уровнем рождаемости; низким уровнем миграций. Молекулярной основой заболевания является увеличение у якутов числа тринуклеотидных CAG-повторов до 39-71 по сравнению с 19-36 в норме в гене SCA1, локализованного в области 6p22 – p23. Среди различных форма наследственных атаксий в Якутии СЦА 1 встречается в 88,1% всех семей.
- Кучер А.Н., Данилова A.JL, Конева Л.A., Максимова Н.Р., Ноговицина А.Н. Генетико-демографическое изучение народонаселения Республики Саха (Якутия) // Якутский медицинский журнал. – 2005. № 2(10). – С. 4-12.
- Конева Л. A., Кучер А.Н., Максимова Н.Р., Пузырёв В.П. Подходы к моделированию распространенности спиноцеребеллярной атаксии I типа в изолированной популяции // Генетика человека и патология: Сб. науч. трудов / Под ред. В.П. Пузырева. – Вып. 7. – Томск: Печатная мануфактура, 2004. -С. 92-101.
- Конева Л .А., Кучер А.Н., Пузырев В.П., Ноговицына А.Н., Максимова Н.Р., Сухомясова А.Л., Данилова А.Л. Демографические и клинико-генетические особенности распространенности спиноцеребеллярной атаксии I типа в Усть-Алданском и Абыйском улусах республики Саха (Якутия) / Матер, на-уч.-практич. конф. «Актуальные вопросы профилактической медицины». -Улан-Удэ. – 2005. – С. 97-100.
- Конева Л.А., Кучер А.Н., Пузырёв В.П., Максимова Н.Р., Ноговицина А.Н., Сухомясова А.Л., Данилова А.Л., Платонов Ф.А., Коротов М.Н. Характеристика заболеваемости спиноцеребеллярной атаксией I типа в Усть-Алданском и Абыйском улусах Республики Саха (Якутия) / Матер. Между-нар. науч.-практич. конф. «Генетические аспекты патологии человека. Проблемы сохранения генофонда коренных народов Севера», Якутск: НИПК «Сахаполиграфиздат». – 2005. – С. 99-100.
- Конева Л.А., Кучер А.Н., Пузырёв В.П., Ноговицина А.Н., Максимова Н.Р., Сухомясова А.Л., Данилова А.Л. Распространенность спиноцеребеллярной атаксии I типа в Усть-Алданском и Абыйском улусах Республики Саха (Якутия) / Матер. Итоговой науч.-практ. конф. ГУ НИИ медицинских проблем севера СО РАМН «Вопросы сохранения и развития здоровья населения Севера и Сибири», Красноярск. – 2005. – С. 127-129.
- Конева Л.А., Максимова Н.Р. Распространенность спиноцеребеллярной атаксии I типа в Якутии: проблемы и пути решения / Матер, конкурса работ молодых ученых «Теоретические и прикладные проблемы медицинской генетики» СО РАМН, Новосибирск. – 2004. – С.103-109.
- Конева Л .А., Максимова Н.Р., Кучер А.Н., Пузырёв В.П. Динамика частоты спиноцеребеллярной атаксии I типа у якутов с учетом специфики популяционной структуры / Генетика в XXI веке: современное состояние и перспективы развития: Матер. 3-го Съезда ВОГиС. -М. – 2004. – С.28.

